Personal Care
Revolutionizing healthcare: The world’s first USDA certified organic OTC medicines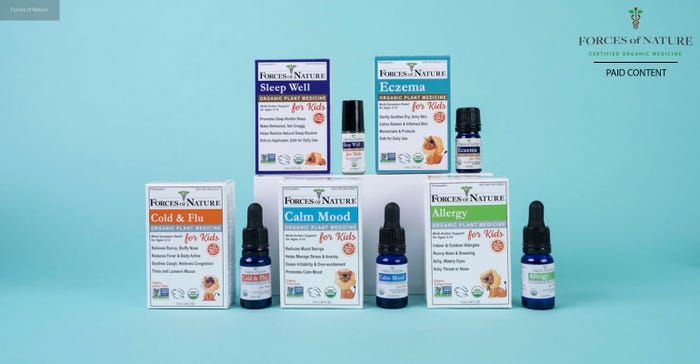
Sponsored ContentRevolutionizing healthcare: The world’s first USDA certified organic OTC medicines – press releaseRevolutionizing healthcare: The world’s first USDA certified organic OTC medicines – press release
Forces of Nature is a leading provider of organic healthcare solutions, dedicated to offering safe and effective remedies for common health concerns.








































Featured


Subscribe and receive the latest updates on trends, data, events and more.
Join 57,000+ members of the natural products community.